
Transforming Neurodegenerative Disease Research With iPSCs: A Path To Innovation And Therapeutic Breakthroughs
By Booma Yandava, MS, MBA

Neurodegenerative disorders (NDs) pose a significant challenge for medicine and due to global aging population, global demographic changes and the serious or life-threatening nature of the disease themselves. Many of these diseases are genetic, although other causes such as toxins, chemicals, and alcoholism have not been ruled out.1,2
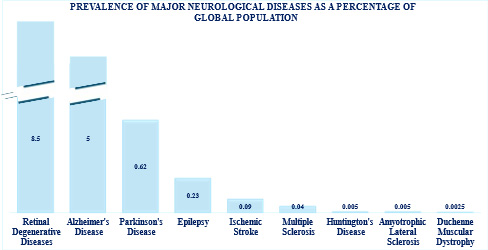
Figure 1: Prevalence in percentage of major neurological disorders by factors globally from 1990 to 2021 https://doi.org/10.1016/ S1474-4422(24)00038-3
At the cellular level, NDs are commonly associated with gradual loss of neurons and synaptic connections particularly in the later phases of a patient’s life. Often the degree of neurological loss directly correlates with the appearance of and progression of clinical symptoms. These disorders ranked second for global prevalence with a total of about 10 million deaths and close to 350 million affected individuals worldwide3 (Figure 1).
Therapeutic Approaches
Despite advances in neuroprotective agents and gene-based therapies, current treatments for neurological disorders primarily manage symptoms rather than address the underlying pathology. This limitation arises from the nonregenerative nature of central nervous system neurons, the complexity of disease mechanisms, challenges in target validation, and the lack of translatable models and reliable biomarkers. Given these obstacles, regenerative approaches, particularly stem cell-based therapies, have emerged as promising alternatives. Among them, induced pluripotent stem cells (iPSCs) stand out due to their unlimited self-renewal capacity, ability to differentiate into neural lineages, and potential for both disease modeling and therapeutic applications. This article focuses on the latest developments in applying iPSC technology to neurological diseases, highlighting its promise, ongoing clinical advancements, and the challenges that must be addressed for successful translation into clinical practice.4
Inside Induced Pluripotent Stem Cells
In 2006, Yamanaka and Takahashi5 demonstrated that introducing these four specific genes that encode for transcription factors Oct3/4, Sox2, Klf4, and c-Myc, somatic cells could be converted to pluripotent cells, that essentially behave like ESCs but originate from any normal tissue such as endothelial cells that further can differentiate into multi-lineage primary germ layers (endoderm, mesoderm, and ectoderm). These reprogrammed cells, or iPSCs, have become a central focus in contemporary regenerative medicine and specifically for neurological disorders.
Recent clinical studies have increasingly favored iPSCs over embryonic stem cells (ESCs) primarily due to their distinct advantages related to ethical, practical, and scientific considerations. iPSCs are generated by reprogramming somatic cells, thus circumventing ethical controversies associated with the use of embryos required for ESC derivation. Furthermore, autologous iPSCs offer the significant benefit of enabling patient-specific cell generation, minimizing the risk of immune rejection and enhancing the potential for personalized therapies. Additionally, iPSCs uniquely facilitate patient-specific disease modeling, allowing researchers to investigate disease pathophysiology, assess therapeutic responsiveness in a personalized context, and perform precision medicine studies that are difficult or impossible with ESCs. Consequently, iPSCs have emerged as the preferred tool in contemporary clinical trials and translational research in regenerative medicine and neurological disorders.
Clinical Landscape of Neurological Disorders with iPSCs
Neurological disorders currently represent the largest share of ongoing iPSC-based clinical trials, comprising approximately 28% of the total (Figure 1, left panel). This underscores the heightened interest and promising potential of iPSCs in addressing complex neurological pathologies.
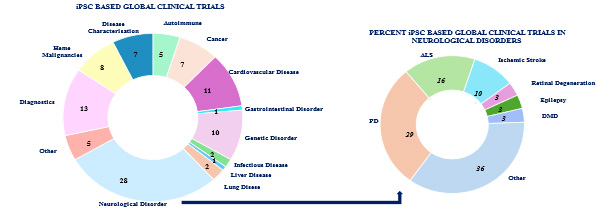
Figure 2: The current landscape of clinical trials utilizing iPSC across neurological disorders has expanded significantly, covering a broad spectrum of medical indications. Source: Clinical trials.gov (2025) and publications
Among the neurological disorders (Figure 2, right panel), Parkinson’s disease (PD) emerges as a primary target, accounting for approximately 29% of neurological iPSC trials. Amyotrophic lateral sclerosis (ALS) is also prominently represented, comprising roughly 16% of these trials. Additionally, indications such as ischemic stroke, epilepsy, retinal degeneration, and Duchenne muscular dystrophy (DMD) demonstrate the broad applicability of iPSC technology in neurological therapeutic research.
Collectively, these figures highlight the growing translational momentum of iPSC-based approaches, particularly in neurodegenerative and neurological disorders, underscoring the significant clinical interest and ongoing efforts to harness the therapeutic potential of this promising technology.
Parkinson's Disease
Parkinson's disease (PD) represents one of the most common neurodegenerative disorders, characterized by progressive loss of dopaminergic neurons in the substantia nigra. Current pharmacological approaches primarily manage symptoms without addressing the underlying neurodegeneration. Cell replacement therapy using iPSCs has emerged as a promising approach for restoring dopaminergic function and potentially modifying disease progression.
The first iPSC based clinical trial for PD was developed by Doi et al.6 at the Center for iPS Cell Research and Application (CiRA) in Japan, established a protocol for producing mesencephalic dopaminergic(mesDA) neurons from stem cells and demonstrated the safety of autologous iPSC derived dopaminergic neuronal cells. This trial is further expanded in the USA (NCT06482268). CORIN sorting strategy is a milestone in the derivation of an improved protocol and in eliminating tumorigenesis. This improvement also resulted in removing serotonin neurons and precursors which are known to result in dyskinesia when transplanted into patients.
Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS) is a lethal, progressive neurodegenerative disease characterized by motor neuron atrophy, gliosis, and the accumulation of misfolded proteins aggregates, such as TAR DNA-binding protein 43 (TDP-43) dipeptide repeat proteins and fused in sarcoma (FUS) protein mislocation. Although, more than 30 at-risk genes have been identified to indicate the pathogenesis of ALS,7 the mechanisms and specific causative genes have not yet been identified. Further, there is considerable heterogeneity in the phenotypes posing challenges in understanding the etiology and development of treatment.
Although clinical trials have been conducted using ESC-derived astrocytes (AstroRx, NCT03482050), there were no significant improvements beyond baseline at 12 months of follow-on studies . Current clinical trials listed the ClinicalTrials.gov are in the early stages of development of iPSCs from ALS patients. iPSCs can be derived from ALS patients with various genotypes and phenotypes, maintaining the genetic background of the donor cells during reprogramming.
The ‘Answer ALS’ initiative has successfully developed a comprehensive repository of iPSCs derived from patients with amyotrophic lateral sclerosis (ALS), offering researchers an unprecedented resource to investigate disease mechanisms, identify novel therapeutic targets, and facilitate personalized treatment strategies.
Ischemic Stroke
Ischemic Stroke is a multifactorial central nervous system (CNS) disorder with a high mortality and morbidity rate and is a result of and is a result of cancer and myocardial infarction. Almost 80% of stroke cases are ischemic stroke, and the remaining are hemorrhagic stroke. Although thrombolysis and thrombectomy are the standard of care and improved prognosis in selected patients, in the majority of patients brain tissue ischemia can cause irreversible damage to neural tissues, leading to persistent neurological dysfunction and the loss of nerve cells. Therefore, novel therapeutic strategies based on regenerative medicines using cell-based therapies are highly desired.
Various non-neural lineage cells including mesenchymal stem cells (MSCs), CD34-positive hematopoietic stem cells (HSCs), umbilical cord blood (UCB), and bone marrow (BM)-derived mononuclear cells (MNCs) as well as neural progenitor cells (NSPCs) isolated from neural tissues neural and progenitor cells (NPCs) differentiated from embryonic stem cells (ESCs) and iPSCs have been investigated as sources for cell-based therapies for ischemic stroke.7 Currently the clinicaltrials.gov lists three trials that are under phase 1 study. Hopstem Biotechnology Inc.is evaluating human forebrain neural progenitor cells (hNPC01) for chronic ischemic stroke. Guidon Pharmaceutics Ltd. Is studying safety and preliminary efficacy of intravenous exosomes derived from human iPSC (GD-iExo-003) in acute ischemic stroke and Allife Medical Science and Technology Co., Ltd. Is conducting efficacy studies of allogeneic endothelial progenitor Cells (EPCs) in Patients with Acute Ischemic Stroke and the results are awaited.
In a pre-clinical study conducted by Kanemura Y.8 and his team with a clinical-grade iPSC derived neural progenitor cells from donors who were homozygous for human leukocyte antigen (HLAs) and tested in rat models, the researchers observed that although, in vivo survival, differentiation and cell replacement may be observed, their efficacy is insufficient for neurological functional improvement after acute or subacute transplantation.
These results suggest the need for further research and optimization of conditions to address the efficacy and safety of iPSC-based therapies in clinical settings.
Retinal Degenerative Diseases
Retinal degenerative diseases represent a group of progressive conditions characterized by photoreceptor and retinal pigment epithelium (RPE) cell death, leading to irreversible vision loss and blindness. These represent one of the most common forms of sensory neurodegeneration and are a significant global health burden affecting millions worldwide. These include age-related macular degeneration (AMD), retinitis pigmentosa, Stargardt disease, and diabetic retinopathy, which collectively affect millions worldwide. Traditional treatments primarily manage symptoms or slow progression but cannot replace lost retinal cells. iPSC-based approaches offer several advantages: they provide a renewable source of patient-specific or HLA-matched retinal cells, avoiding ethical concerns associated with embryonic stem cells; they can be genetically corrected before differentiation when treating inherited disorders; they can be expanded extensively to meet clinical demands.
Mandai, M. et al.9 were the first ones to demonstrate the feasibility of iPSC-based approach for age-related macular degeneration (AMD). Autologous iPSC-derived retinal pigment epithelium (RPE) sheets were transplanted and One patient showed stabilized vision without serious adverse events, but high production costs and genetic abnormalities in a second patient's cells led to protocol modification. A subsequent trial with transplanted iPSC derived RPE sheets in a patient with AMD resulted in graft survival even though visual acuity did not improve significantly. In an allogeneic iPSC-derived RPE cells transplantation study, RIKEN, five patients received transplants with no serious adverse events; modest improvements in visual function were reported in 60% of patients.
Notable on-going clinical trials showing promise include - National Eye Institute (NEI)/NIH clinical trial with Autologous iPSC-derived RPE patch for AMD. Transplantation of iPSC-derived photoreceptor progenitor cells for retinitis pigmentosa demonstrated safety with no tumor formation or immune rejection by Fate therapeutics. This trial showed modest improvements in light perception in 2/6 patients. Off-the-shelf iPSC-derived RPE cells for geographic atrophy by Lineage Cell Therapeutics with early data suggesting slowing of disease progression and iPSC-derived retinal organoids for advanced retinitis pigmentosa by Lineage Cell Therapeutics where some patients showed stabilization of visual field loss.
Much anticipated results are from ReNeuron/Fosun Pharma whose approach is based on iPSC-derived 3D retinal tissue with integrated vasculature and Sumitomo Dainippon Pharma/CiRA ‘s technology whose CRISPR-edited allogeneic iPSC-derived photoreceptors diseases showed no severe immune reactions in early Phase 1 and efficacy assessment is anticipated.
Epilepsy
Epilepsy is a neurological condition defined by an enduring predisposition to experience epileptic seizures, accompanied by significant neurobiological, cognitive, psychological, and social implications.10 The persistent exposure to unpredictable seizure events along with associated comorbidities, treatment regimens, and their collective impact on individuals' daily functioning imposes major elements of epilepsy's burden. These factors have led to conceptualization of epilepsy within a chronic stress framework in human studies though the relationship between stress and epilepsy exhibits considerable complexity.
As of now, there are no registered clinical trials employing iPSC-based therapies specifically for epilepsy. However, Disease Modeling Studies are actively exploring the potential of iPSCs in modeling epilepsy and developing therapeutic interventions.
iPSC-derived models have significantly advanced epilepsy research by enabling patient-specific investigations of neuronal dysfunction and drug screening. Liu Y. et al.11 used iPSC-derived neurons from Dravet syndrome patients to reveal Nav1.1 channel dysfunction and neuronal hyperexcitability, validating cannabidiol’s therapeutic effects. Further iPSC-derived cerebral organoids were developed from PCDH19 epilepsy patients, demonstrating impaired network synchronization and identifying compounds that restore calcium signaling. Similarly, Nadadhur A.G et al.12 generated iPSC-derived cortical networks from Tuberous Sclerosis Complex (TSC) patients, showing that these models accurately predicted responses to mTOR inhibitors than traditional methods, enhancing precision medicine approaches. Extracellular vehicles (EVs) isolated from GABAergic interneurons generated from iPSCs were demonstrated to regulate seizures in temporal lobe epilepsy. It is identified that the cellular origin of EVs plays a vital role in seizure control with exogenous GABA.13 These studies highlight iPSC-based approaches as promising tools for both disease modeling and therapeutic intervention in epilepsy.
Muscular Dystrophy
Duchenne muscular dystrophy (DMD) is the most prevalent form of muscular dystrophy, affecting approximately 1 in 3,500 live-born males due to its X-linked recessive inheritance pattern. The disease arises from mutations in the DMD gene (Xp21.2–p21.1), which encodes dystrophin, a structural protein essential for maintaining the integrity of striated muscle fibers. Loss of dystrophin disrupts muscle cell stability, leading to progressive degeneration of skeletal and cardiac muscle. Clinically, DMD manifests as progressive muscle weakness and atrophy, with affected individuals typically experiencing loss of ambulation by age 12. Despite advances in research, DMD currently lacks curative treatment.14
iPSCs have become an invaluable platform for modeling muscular dystrophies and facilitating drug discovery. Maffioletti et al.15 developed a three-dimensional artificial skeletal muscle model from iPSC-derived myogenic cells of Duchenne muscular dystrophy (DMD) patients, revealing structural abnormalities and calcium handling defects. This system provided a robust platform for testing dystrophin-restoring compounds. Further, Metzler et al.16 showed that the generation of muscle-stem-derived iPSCs is a fast and economic method to obtain unrestricted cell numbers for cell-based therapies in muscle wasting disorders, and in this aspect are superior to blood-derived iPSCs.
Further advancing high-throughput drug screening approaches, Ueki et al.17 developed an iPSC-based platform for myotonic dystrophy type 1, successfully identifying small molecules that correct RNA splicing defects in patient-derived cells. CRISPR-Cas9 has been reported as an efficient tool for inducing exon skipping in iPSCs 5 and in vivo animal DMD models 6–9 to restore dystrophin protein expression.18
The translation of iPSC-based therapies into clinical applications is promising yet preliminary. A Phase I safety trial involving iPSC-derived myogenic progenitor cell transplantation in seven DMD patients, reporting no serious adverse effects and modest dystrophin expression in five patients at biopsy sites. A Phase I/II trial, investigated genetically corrected autologous iPSC-derived myogenic cells in limb-girdle muscular dystrophy, demonstrating safety and localized functional improvement in three out of eight patients. iPSC-derived meso-angioblast-like cells for DMD, confirmed safety and dystrophin expression but limited functional recovery. More recently, clinical trials are being initiated employing CRISPR-corrected iPSC-derived muscle stem cells for DMD, with interim results showing successful engraftment without major complications, highlighting the potential of gene-corrected iPSC-based therapies in muscle regeneration.
Challenges, Opportunities and Future Directions
The use of iPSCs in neurological disease research and therapy represents a groundbreaking advancement in regenerative medicine. By offering a patient-specific, pluripotent platform, iPSCs enable disease modeling, drug discovery, and potential cell-based therapies. However, despite these promising applications, several critical challenges must be addressed before iPSC-derived therapies can achieve widespread clinical translation.
One of the primary hurdles in applying iPSCs to neurological diseases is the efficient and reproducible differentiation into functional neural subtypes, such as dopaminergic neurons (for Parkinson’s disease) or motor neurons (for ALS). Despite advances in differentiation protocols, iPSC-derived neurons often exhibit immature phenotypes raising concerns regarding functional integration and long-term stability. iPSCs carries the risk of genomic instability and tumorigenicity due to potential mutations, epigenetic alterations, and incomplete reprogramming. Although autologous iPSCs theoretically reduce immune rejection, in practice, epigenetic reprogramming and prolonged in vitro culture may alter self-recognition, triggering unexpected immune responses.
However, the integration of CRISPR-Cas9 gene editing with iPSC technology has enabled the correction of disease-causing mutations, paving the way for patient-specific, genetically optimized neural cells. This approach holds immense promise for disorders such as Huntington’s disease, ALS, and certain forms of hereditary Parkinson’s disease, where correcting genetic defects at the cellular level could prevent disease progression. The development of iPSC-derived brain organoids and 3D neural cultures provide unprecedented insights into disease mechanisms, drug testing, and personalized medicine. The development of allogeneic iPSC banks from immunologically diverse donors offers a promising solution to immune rejection concerns. Efforts are underway to establish disease-specific iPS cell lines repositories from hereditary neurological disease patients. The cell lines will be registered and make them available to other investigators.
Conclusions
In conclusion, neurological disorders have a major impact on families and the national health service due to the lack in many cases of effective and long-lasting therapies. The lack of these therapeutic strategies is due in large part to the difficulty of modeling these pathologies in vitro. The emerging domain of iPSC-based therapeutic approaches represents an equilibrium between potential and obstacles. Early-stage clinical studies have paved the way for the translation of iPSC-based therapies from preclinical to clinical trials demonstrating safety and potential efficacy of iPSC derived cell transplantation. Ongoing clinical studies aim to validate these findings in larger cohorts and over extended follow-up periods to establish long-term outcomes. Similarly, iPSC-based pre-clinical approaches demonstrate promising early clinical results following extensive disease modeling. While these therapies remain experimental, they represent significant advances in addressing conditions with limited treatment options. The coming years will be critical in determining their long-term efficacy and safety profiles.
About the Author
Booma Yandava, MS, MBA, PhD (2026)
 Booma Yandava is a biotechnology executive and thought leader specializing in venture capital investments, strategic partnerships, and biopharmaceutical innovation. With extensive expertise in drug development, licensing transactions, and commercialization strategies, she has played a pivotal role in shaping business and investment strategies for biotechnology companies. Her work focuses on oncology, rare diseases, and neurological disorders, guiding decision-making at the intersection of science, business, and investment.
Booma Yandava is a biotechnology executive and thought leader specializing in venture capital investments, strategic partnerships, and biopharmaceutical innovation. With extensive expertise in drug development, licensing transactions, and commercialization strategies, she has played a pivotal role in shaping business and investment strategies for biotechnology companies. Her work focuses on oncology, rare diseases, and neurological disorders, guiding decision-making at the intersection of science, business, and investment.
Yandava’s research explores risk mitigation and long-term value creation in biopharma venture capital, leveraging her background in technology transfer, financial strategy, and market intelligence to inform investment and partnership decisions. She has contributed to peer-reviewed publications, presented at leading industry conferences, and provided strategic insights on emerging trends in cell and gene therapies, regenerative medicine, and biopharma deal-making.
Bibliography
- Kim, M. O., & Geschwind, M. D. (2018). Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harbor perspectives in biology, 10(4), a033118
- Collaborators, G. B. D. (2019). Mental Disorders, et al.(2022) Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: A systematic analysis for the global burden of disease study 2019. The Lancet Psychiatry, 9(2), 137-150.
- Dugger BN, Dickson DW. (2017). Pathology of neurodegenerative diseases. Cold Spring Harbor Perspectives in Biology, 9(7), a028035.
- Cummings J. (2022). New approaches to disease modifying therapies for neurodegenerative disorders. Journal of Prevention of Alzheimer's Disease, 9(2), 280-287
- Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell, 126(4), 663-676.
- Doi, D., Magotani, H., Kikuchi, T., Ikeda, M., Hiramatsu, S., Yoshida, K., ... & Takahashi, J. (2020). Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nature communications, 11(1), 3369
- Marei, H. E., Hasan, A., Rizzi, R., Althani, A., Afifi, N., Cenciarelli, C., ... & Shuaib, A. (2018). Potential of stem cell-based therapy for ischemic stroke. Frontiers in neurology, 9, 271733
- Kanemura, Y., Yamamoto, A., Katsuma, A., Fukusumi, H., Shofuda, T., Kanematsu, D., ... & Okano, H. (2024). Human-induced pluripotent stem cell-Derived neural progenitor cells showed neuronal differentiation, neurite extension, and formation of synaptic structures in rodent ischemic stroke brains. Cells, 13(8), 671
- Mandai, M., Watanabe, A., Kurimoto, Y., Hirami, Y., Morinaga, C., Daimon, T., ... & Takahashi, M. (2017). Autologous induced stem-cell–derived retinal cells for macular degeneration. New England Journal of Medicine, 376(11), 1038-1046
- Liu, X., Sun, X., Sun, C., Zou, M., Chen, Y., Huang, J., ... & Chen, W. X. (2022). Prevalence of epilepsy in autism spectrum disorders: A systematic review and meta-analysis. Autism, 26(1), 33-50
- Liu, Y., Lopez‐Santiago, L. F., Yuan, Y., Jones, J. M., Zhang, H., O'Malley, H. A., ... & Parent, J. M. (2013). Dravet syndrome patient‐derived neurons suggest a novel epilepsy mechanism. Annals of neurology, 74(1), 128-139.
- Nadadhur, A. G., Alsaqati, M., Gasparotto, L., Cornelissen-Steijger, P., van Hugte, E., Dooves, S., ... & Heine, V. M. (2019). Neuron-glia interactions increase neuronal phenotypes in tuberous sclerosis complex patient iPSC-derived models. Stem cell reports, 12(1), 42-56.
- Chandran, D., Hegde, S., Upadhya, R., SE, P. K., Shenoy, S., Devi, V., & Upadhya, D. (2024). Cell-specific extracellular vesicle-encapsulated exogenous GABA controls seizures in epilepsy. Stem Cell Research & Therapy, 15(1), 1-15.
- Crisafulli, S., Sultana, J., Fontana, A., Salvo, F., Messina, S., & Trifirò, G. (2020). Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet journal of rare diseases, 15, 1-20.
- Maffioletti, S. M., Sarcar, S., Henderson, A. B., Mannhardt, I., Pinton, L., Moyle, L. A., ... & Tedesco, F. S. (2018). Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Reports, 23(3), 899-908.
- Metzler, E., Escobar, H., Sunaga-Franze, D. Y., Sauer, S., Diecke, S., & Spuler, S. (2022). Generation of hiPSC-derived skeletal muscle cells: exploiting the potential of skeletal muscle-derived hiPSCs. Biomedicines, 10(5), 1204.
- Ueki, J., Nakamori, M., Nakamura, M., Nishikawa, M., Yoshida, Y., Tanaka, A., ... & Sakurai, H. (2017). Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Scientific reports, 7(1), 42522.
- Wang, P., Li, H., Zhu, M., Han, R. Y., Guo, S., & Han, R. (2023). Correction of DMD in human iPSC-derived cardiomyocytes by base-editing-induced exon skipping. Molecular Therapy Methods & Clinical Development, 28, 40-50.